암 세포주의 유전자 발현 데이터를 이용하여 특정 약물에 대한 반응(AUC, IC50)을 예측하는 모델을 만듭니다. 정밀의학 기반의 개인맞춤 치료 후보 약물 도출에 응용 가능합니다.
📂 필요한 라이브러리

💻 Python 예시 코드

import pandas as pd
from sklearn.ensemble import RandomForestRegressor
from sklearn.model_selection import train_test_split
from sklearn.metrics import mean_squared_error
# 가상의 유전자 발현 + 약물 반응 데이터
df = pd.read_csv("drug_response.csv") # features: gene1~geneN, target: IC50
X = df.drop(columns=["IC50"])
y = df["IC50"]
X_train, X_test, y_train, y_test = train_test_split(X, y, test_size=0.2)
model = RandomForestRegressor()
model.fit(X_train, y_train)
y_pred = model.predict(X_test)
print("RMSE:", mean_squared_error(y_test, y_pred, squared=False))
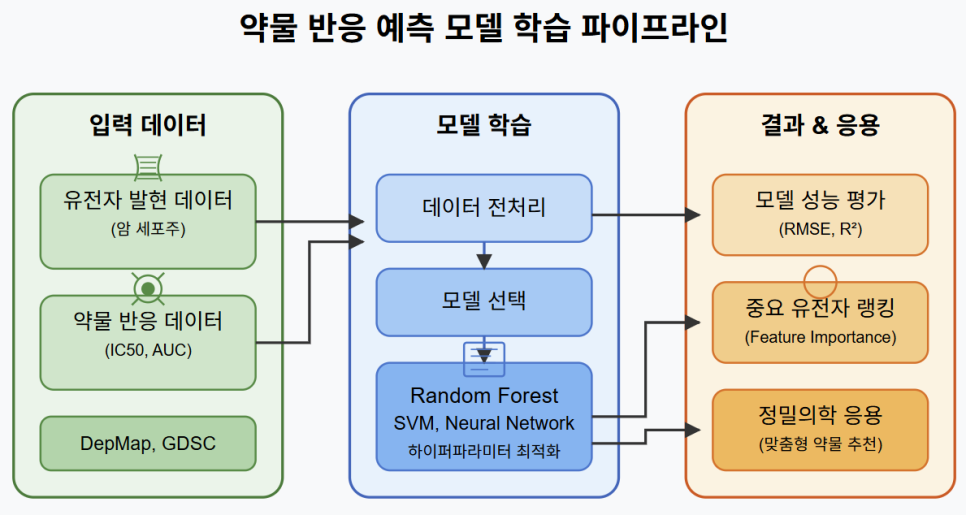
그림 3. 약물 반응 예측 모델 학습 파이프라인
- 입력: 유전자 발현 행렬 + IC50
- 처리: 모델 학습 (Random Forest 등)
- 출력: 예측 IC50, 중요 유전자 ranking
🔗 관련 링크
- DepMap Portal
- GDSC Database
- DeepChem (약물-유전체 예측용)
🧬 이론적 배경
- 암세포는 유전자 발현 프로파일에 따라 특정 약물에 민감하거나 저항성을 보입니다. 이 정보를 활용해 개인 맞춤형 약물 처방 전략을 개발하는 것이 정밀의학의 핵심입니다.
- GDSC, CCLE, DepMap과 같은 데이터베이스는 수백 개 세포주에 대해 유전자 발현과 약물 반응(AUC, IC50) 데이터를 제공합니다.
- 머신러닝 회귀 모델은 이런 다변량 데이터를 학습하여, 유사한 발현 패턴을 가진 신규 샘플에 대해 반응 값을 예측할 수 있습니다.

import pandas as pd
from sklearn.model_selection import train_test_split
from sklearn.ensemble import RandomForestRegressor
from sklearn.metrics import mean_squared_error
# Load dataset: columns are gene1, gene2, ..., IC50
df = pd.read_csv("drug_response.csv") # Replace with actual dataset
# Features: gene expression data, Target: IC50
X = df.drop(columns=["IC50"])
y = df["IC50"]
# Train-test split
X_train, X_test, y_train, y_test = train_test_split(X, y, test_size=0.2, random_state=42)
# Train Random Forest model
model = RandomForestRegressor(n_estimators=100)
model.fit(X_train, y_train)
# Predict and evaluate
y_pred = model.predict(X_test)
rmse = mean_squared_error(y_test, y_pred, squared=False)
print(f"Test RMSE: {rmse:.3f}")
🔍 분석 개요
- 입력 변수는 세포주의 유전자 발현 데이터이며, 목표 변수는 특정 약물에 대한 IC50 값(또는 AUC 등)입니다. 이 문제는 다변량 회귀 문제로 정의할 수 있습니다.
- 일반적으로 Random Forest, XGBoost, ElasticNet, Deep Neural Network 등이 많이 사용되며, 성능 비교를 위해 다양한 모델을 시도해 보는 것이 좋습니다.
- 모델이 학습된 후, 실제 실험되지 않은 세포주에 대해 가상 약물 반응을 예측하거나, 반응이 좋을 것으로 예측된 상위 약물을 도출할 수 있습니다.

import shap
# Initialize SHAP explainer for tree-based model
explainer = shap.Explainer(model, X_test)
# Compute SHAP values for the first 50 samples
shap_values = explainer(X_test[:50])
# Plot summary of feature importance (global view)
shap.plots.beeswarm(shap_values)
⚠️ 주의할 점
- 유전자 수는 수천 개에 달하지만 세포주의 수는 수백 개로 제한되기 때문에, 차원 축소나 주요 유전자 선택을 하지 않으면 과적합이 발생하기 쉽습니다.
- 약물별로 반응 분포가 매우 다르기 때문에, 약물별로 별도 모델을 학습하거나, 반응 분포 정규화를 고려하는 것이 좋습니다.
- 실험값(IC50 등)의 단위와 스케일이 모델 성능에 영향을 줄 수 있으므로, 반드시 정규화 혹은 로그 변환을 검토해야 합니다.
➕ 추가 분석 가능한 내용
- SHAP 값을 이용하면 모델이 예측에 사용하는 주요 유전자들의 상대적 중요도를 시각적으로 파악할 수 있어 해석력이 높아집니다.
- 단일 약물 예측을 넘어 다약물 예측 또는 세포주 간 약물 선택 최적화와 같은 멀티태스크 모델도 실험할 수 있습니다.
- 약물의 타깃 단백질 정보나 구조 정보(화학 fingerprint)를 함께 사용하여 멀티모달 모델로 확장할 수 있습니다.